

Changes in DNA methylation are characteristic for patients with acute myeloid leukemia (AML) and many studies have reported the prognostic significance of these epigenetic aberrations. We aimed for a complex evaluation of DNA methylation changes in AML patients at diagnosis. Therefore, we designed a sequencing panel targeting 239 regions annotated to 186 genes previously described in literature as having a prognostic impact or being commonly associated with AML pathogenesis (e.g. WT1, HOX genes).
We used diagnostic whole-blood DNA samples of adult AML patients who were treated with curative intent starting with 3+7 induction regimen. In the testing cohort, we sequenced 128 AML patients and 11 samples from healthy donors. We analyzed another 50 AML patients from partner institution University Hospital Brno as an independent validation cohort. The libraries were prepared using SeqCap Epi Enrichments System (Roche) and sequenced on MiSeq instrument (Illumina). Data were processed in Linux opensource software and further analyzed in R.
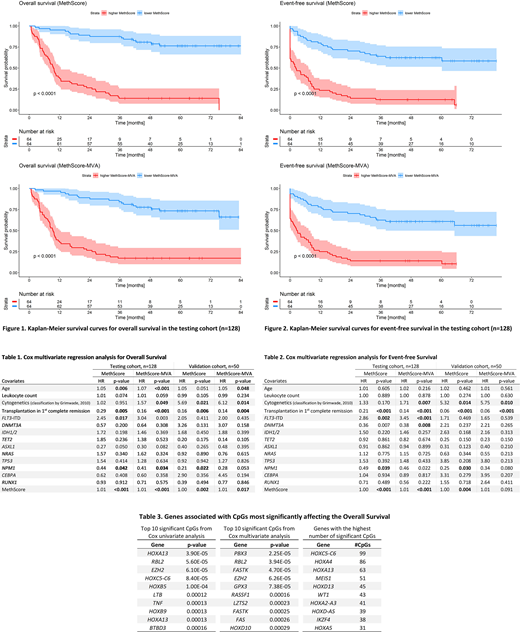
For each sample, we measured the methylation level of nearly 50 000 CpGs. In the testing cohort, we used the Cox regression to evaluate the effect of each CpG's methylation level on overall survival (OS). As a result, we found 1961 CpGs significantly effecting the OS (p<0.05) annotated to 141 genes. In gene ontology analysis, these loci were mainly connected to transcription and RNA regulation, DNA binding, and embryonic development. In 1097 CpGs, higher methylation indicated better outcome and, on the contrary, a poorer prognosis in the remaining 864 CpGs. Next, we used linear combination of the methylation levels and Cox's beta regression coefficients for each CpG to count a summarizing value that we called MethScore. Patients with lower MethScore (n=64) had markedly longer OS and event-free survival (EFS) than patients with higher MethScore (n=64, Logrank test for OS: p<2e-16, for EFS: p<2e-12). To further specify the prognostically most influential CpGs out of the selected 1961 loci, we subjected them individually to Cox multivariate regression analysis together with age, leukocytes count, cytogenetics, FLT3-ITD mutation, and transplantation in the 1st complete remission. Only 625 CpGs remained significant (p<0.05) and from these, we calculated MethScore-MVA via the same procedure as MethScore. MethScore-MVA was also very well predictive of patients' survival (Logrank test comparing patients with higher and lower MethScore-MVA for OS: p<7e-14, for EFS: p<2e-10). MethScore and MethScore-MVA proved to be the most significant variables in multivariate Cox regression for both OS (p=2e-12 and p=2e-13) and EFS (p=2e-12 and p=3e-12, respectively).
To validate these results, we counted MethScore and MethScore-MVA for 50 patients from the validation cohort and performed the same Cox multivariate analysis. MethScore remained highly significant for both OS and EFS (p=0.002 and p=0.004, respectively). MethScore-MVA was significant for OS (p=0.02) but not for EFS (p=0.09).
Here, we present the MethScore as a novel complex characterization of predictive DNA methylation changes in patients with AML. MethScore might be used as a new surrogate marker that could enrich the currently used cytogenetic and genetic markers to refine the prognostication of newly diagnosed AML patients.
This study was supported by the Ministry of Health of the Czech Republic, project for conceptual development of research organizations (00023736, IHBT).
Šálek:Amgen: Consultancy, Honoraria, Research Funding. Mayer:Celgene: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal